Denovo genome assembly using long reads
Generated by: George Edward Chlipala
Report date: April 25, 2024
Overview
When you publish manuscripts based on data generated at our facility, we would greatly appreciate an acknowledgement of our efforts. Please cite our facility as follows (for example):
Basic processing of the raw data were performed by the University of Illinois at Chicago Research Informatics Core (UICRIC).
We adhere to a general policy for acknowledgements and authorship as established by the Association for Biomolecular Resource Facilities (ABRF) , and we support the following statement from the ABRF.
The existence of core facilities depends in part on proper acknowledgment in publications. This is an important metric of the value of most core facilities. Proper acknowledgment of core facilities enables them to obtain financial and other support so that they may continue to provide their essential services in the best ways possible. It also helps core personnel to advance in their careers, adding to the overall health of the core facility.
Please contact us for assistance in drafting manuscripts.
Output Files
| File | Description | Type |
|---|---|---|
| sample_A.gene-AA.fa | Protein sequences of predicted ORFs for sample_A | result |
| sample_A.gene-NT.fa | Nucleotide sequences of predicted ORFs for sample_A | result |
| sample_A.annotation.txt | Annotations of predicted ORFs for sample_A | result |
| sample_A.gff | Details of predicted ORFs for sample_A in GFF format | result |
| sample_A.gbk | Annotated sequences of contigs for sample_A | result |
| sample_A-contigs.zip | ZIP compressed FASTA file of contigs for sample_A | result |
| Sample | OriginalID |
|---|---|
| sample_A | sample_A |
Details
- Method: FastQC
-
General quality-control metrics for next-generation sequencing data were obtained using FastQC. - Method: FastQC
-
General quality-control metrics for next-generation sequencing data were obtained using FastQC.
Details
- Method: Porechop
-
Custom ParametersAdapter trimmer for Oxford Nanopore reads. - -v = 0
- Method: Minimum length trimming
-
Custom ParametersReads less than specified length were discarded. - length = 1000 bp
Table 1. Trim statistics Download table data
| Sample | Raw reads (reads) | Raw reads (bp) | Passed trimming (reads) | Passed trimming (bp) | Passed length filter (reads) | Passed length filter (bp) |
|---|---|---|---|---|---|---|
| sample_A | 332722 | 815437449 | 331741 | 776727919 | 234373 | 713978446 |
Details
- Method: Flye assembler v2.9
-
Custom ParametersA de novo assembler for single-molecule sequencing reads, such as those produced by PacBio and Oxford Nanopore Technologies. - --asm-coverage = 100
Figure 1. Coverage plot for sample_A
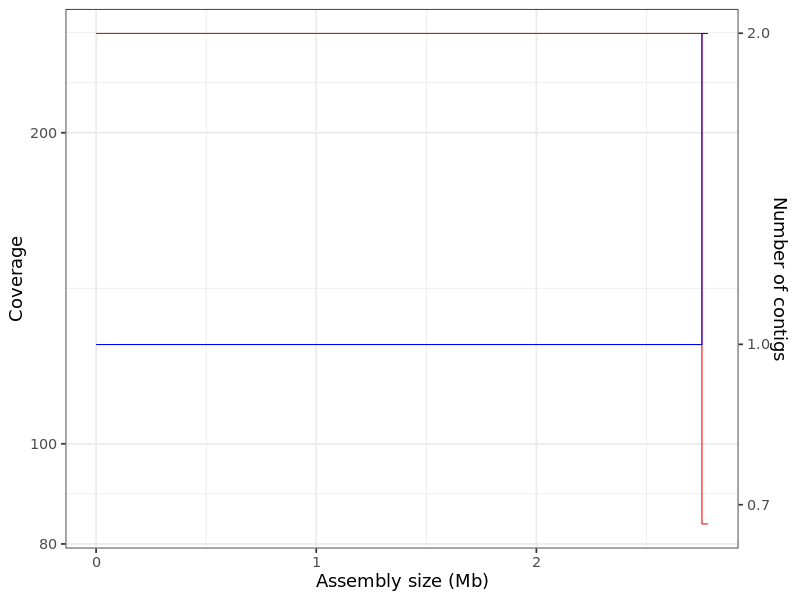
Figure 2. Coverage plot by minimum read length for sample_A
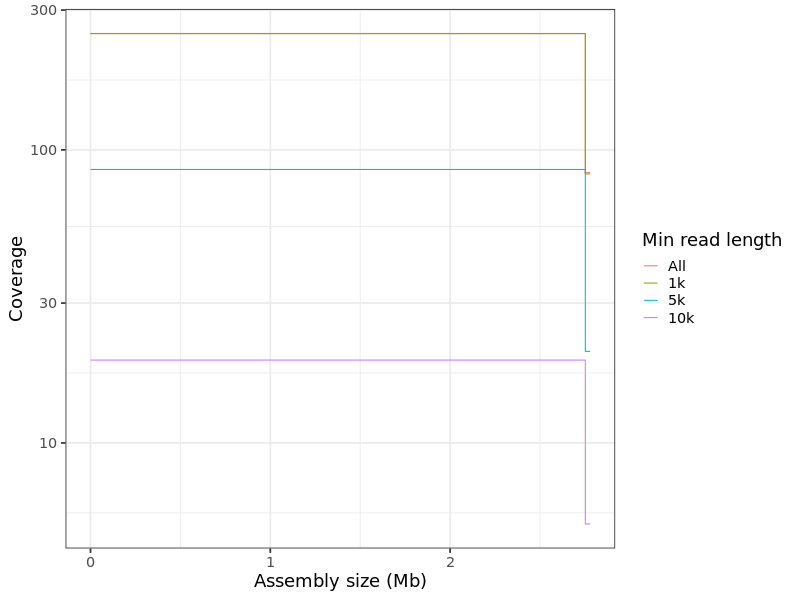
Table 1. Assembly summary for all samples Download table data
| Sample | Target genome size | Target coverage | Count | TotalLength | Longest | N50 | N75 | L50 | L75 |
|---|---|---|---|---|---|---|---|---|---|
| sample_A | 5m | 100 | 2 | 2779040 | 2751939 | 2751939 | 27101 | 1 | 2 |
Table 2. Basic contig statistics for sample_A Download table data
| #seq_name | circ. | repeat | mult. | alt_group | graph_path | length | GC | coverage | C50 | C75 | C90 | cov_read1k | cov_read5k | cov_read10k | S_2mer | S_3mer | S_4mer |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| contig_1 | Y | N | 1 | * | 1 | 2751939 | 32.936 | 249.52 | 243.00 | 170.00 | 117.00 | 249.24 | 85.73 | 19.17 | 0.955 | 0.954 | 0.952 |
| contig_2 | Y | N | 1 | * | 2 | 27101 | 27.316 | 83.64 | 76.00 | 63.00 | 55.00 | 82.78 | 20.53 | 5.29 | 0.921 | 0.921 | 0.919 |
Details
- Method: Naive sequence correction
-
Custom ParametersSequence of contigs were corrected via multiple rounds of mapping read data to contigs with BWA-MEM followed by calling of major variant from resulting sequence pileup. - iter = auto
Figure 1. Comparision of coverage for sample_A
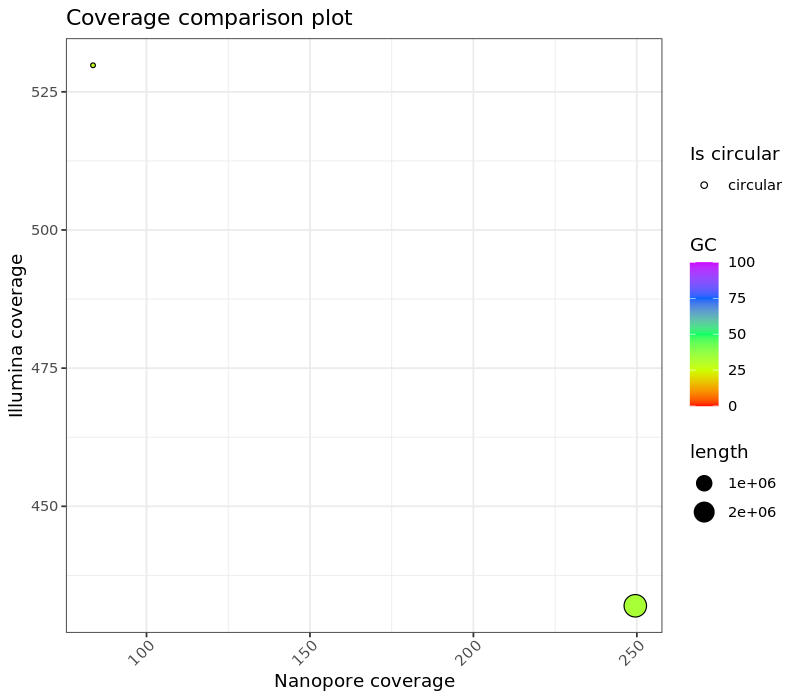
Figure 2. Coverage plot of Illumina data for sample_A
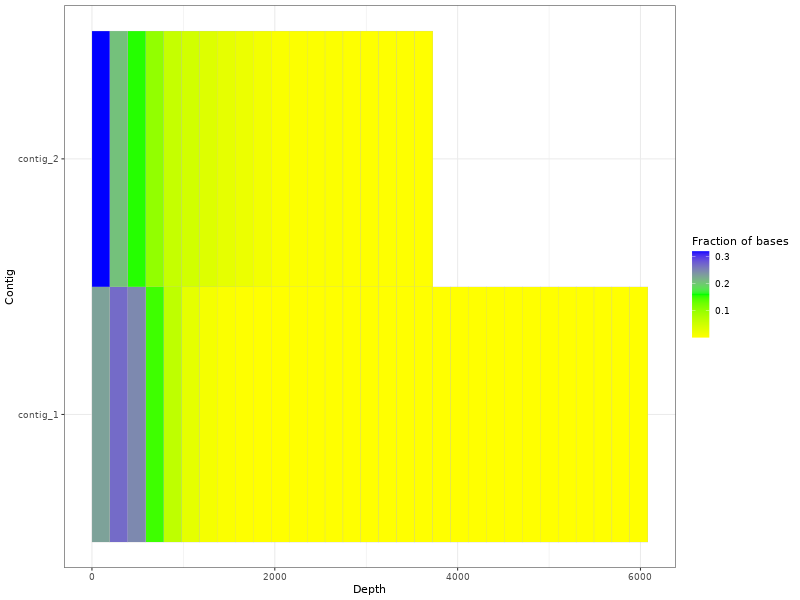
Table 1. Coverage summary of Illumina data for sample_A Download table data
| chromosome | length | avg.coverage | max.cov | min.cov | cov.90 | cov.75 | cov.50 |
|---|---|---|---|---|---|---|---|
| contig_1 | 2760886 | 432 | 5894 | 0 | 93 | 214 | 395 |
| contig_2 | 27269 | 529.8 | 3656 | 18 | 86 | 154 | 368 |
Table 2. Polishing results for sample_A Download table data
| Contig | % IDY | Length raw | Length polished |
|---|---|---|---|
| contig_1 | 99.67 | 2751939 | 2760886 |
| contig_2 | 99.36 | 27101 | 27269 |
Details
- Method: Bakta
-
Custom ParametersRapid & standardized annotation of bacterial genomes, MAGs & plasmids. - --force
Table 1. Annotation details Download table data
| Sample | tRNAs | tmRNAs | rRNAs | ncRNAs | ncRNA regions | CRISPR arrays | CDSs | pseudogenes | hypotheticals | signal peptides | sORFs | gaps | oriCs | oriVs | oriTs | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sample_A | 61 | 1 | 19 | 87 | 25 | 0 | 2537 | 11 | 51 | 0 | 16 | 0 | 5 | 0 | 1 |